

Abstract
Purpose
Alpelisib, a selective oral inhibitor of the class I PI3K catalytic subunit p110α, has shown synergistic anti-tumor activity with endocrine therapy against ER+/PIK3CA mutated breast cancer cells. This phase Ib study evaluated alpelisib plus letrozole’s safety, tolerability and preliminary activity in patients with metastatic ER+ breast cancer refractory to endocrine therapy VSports最新版本.
VSports手机版 - Patients and Methods
Twenty-six patients received letrozole and alpelisib daily. Outcomes were assessed by standard solid-tumor phase I methods. Tumor blocks were collected for DNA extraction and Next Generation Sequencing V体育平台登录.
Results
Alpelisib’s maximum-tolerated dose (MTD) in combination with letrozole was 300 mg/d. Common drug-related adverse events included hyperglycemia, nausea, fatigue, diarrhea and rash with dose-limiting toxicity occurring at 350 mg/d of alpelisib VSports注册入口. The clinical benefit rate (lack of progression ≥6 months) was 35% (44% in patients with PIK3CA mutated and 20% in PIK3CA wild-type tumors; 95% CI [17%; 56%]), including five objective responses. Of eight patients remaining on treatment ≥12 months, six had tumors with a PIK3CA mutation. Among evaluable tumors, those with FGFR1/2 amplification and KRAS and TP53 mutations did not derive clinical benefit. Overexpression of FGFR1 in ER+/PIK3CA mutant breast cancer cells attenuated the response to alpelisib in vitro.
Conclusion
The combination of letrozole and alpelisib was safe, with reversible toxicities. Clinical activity was observed independently of PIK3CA mutation status although clinical benefit was seen in a higher proportion of patients with PIK3CA mutated tumors. Phase II and III trials of alpelisib and endocrine therapy in patients with ER+ breast cancer are ongoing V体育官网入口.
INTRODUCTION
The phosphoinositide 3-kinase (PI3K) pathway is the most frequently altered pathway in cancer, including mutation and/or amplification of the genes encoding the PI3K catalytic subunits p110α (PIK3CA) and p110β (PIK3CB), the PI3K regulatory subunit p85α (PIK3R1), AKT1-3, and the phosphatidylinositol-3,4,5 trisphosphate (PIP3) phosphatases PTEN and INPP4B, among others. PIK3CA mutations induce a transformed phenotype, including growth factor- and anchorage-independent growth, resistance to anoikis and drug resistance(1-4). About 40% of ER+ breast cancers harbor PIK3CA mutations(5-7) VSports在线直播.
We and others have shown that aberrant activation of the PI3K signaling is associated resistance to endocrine therapy(8). PI3K signaling has been shown to promote estrogen-independent growth of ER+ breast cancer cells(9, 10) and this growth is inhibited by the addition of PI3K inhibitors to antiestrogens(11). Additionally, inhibition of PI3K prevents the emergence of hormone-independent cells, which suggests that early intervention with antiestrogens and PI3K inhibitors could limit escape from endocrine therapy V体育2025版.
Drugs targeting multiple levels of the PI3K network have been developed(12, 13) VSports. Alpelisib (BYL719; Novartis Pharma AG, Basel, Switzerland) is an oral inhibitor that selectively targets p110α(14, 15). A phase I study of alpelisib, alone and in combination with fulvestrant, declared its maximum tolerated dose (MTD) as 400 mg/d(16).
The primary objective of this phase Ib trial (NCT01791478) was to determine the safety and tolerability of letrozole, an aromatase inhibitor, in combination with alpelisib in patients with ER+/HER2-negative metastatic breast cancer refractory to endocrine therapies. Secondary objectives included antitumor activity and correlation of clinical outcome with presence of PIK3CA mutations in tumor specimens. Preliminary data from other ongoing clinical trials of alpelisib with letrozole (NCT01872260) show no evidence of pharmacokinetic (PK) drug-drug interactions(17), therefore, no PK analysis was planned for this trial.
PATIENTS AND METHODS
VSports注册入口 - Patient Population
The patient population included post-menopausal patients with histologically confirmed ER+/HER2− metastatic breast cancer refractory to at least one line of endocrine therapy in the metastatic setting, or diagnosed with metastatic breast cancer during or within 1 year of adjuvant endocrine therapy; evaluable disease as defined by Response Evaluation Criteria In Solid Tumors (RECIST); age ≥18 years; life expectancy ≥6 months; ECOG performance status ≤1; adequate bone marrow, hepatic, and renal function; and fasting plasma glucose levels ≤140 mg/dL (7.8 mmol/L). A tumor specimen (primary or metastatic) from archival material or a fresh biopsy was required for study enrollment. Key exclusion criteria were CYP3A4 modifier drug treatment ≤2 weeks before starting alpelisib, clinically-manifested diabetes mellitus, and prior treatment with PI3K inhibitors. Prior treatment with everolimus was allowed.
Approval was obtained from the ethics committees (IRB #101057, Vanderbilt University) at the participating institutions and regulatory authorities. All patients gave informed consent. The study followed the Declaration of Helsinki and Good Clinical Practice guidelines.
Study Design
This phase Ib, multicenter, open-label study enrolled subjects using a standard 3+3 dose escalation design. All patients received letrozole 2.5 mg/d; alpelisib was initiated at 300 mg/d, 25% below the single agent MTD. Both medications were administered daily on a 28-day cycle. In case of adverse events requiring dose adjustments, alpelisib was reduced to 250 mg/d. Intra-patient dose reductions were allowed after the initial 4 weeks of treatment. Patients were treated until disease progression, unacceptable toxicity, or withdrawal of consent.
DLTs were defined as Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 grade ≥3 toxicities. Exceptions were any grade ≥2 toxicity necessitating treatment interruption for more than 21 consecutive days, and non-CTCAE grade 2 hyperglycemia not resolved to grade 0 within 14 consecutive days of initiation of oral anti-diabetic medications (metformin). Grade ≥3 anemia was not considered a DLT unless judged to be a hemolytic process secondary to study drug. Grade ≥3 lymphopenia was not considered a DLT unless clinically significant.
The MTD was defined as the highest dose of alpelisib in combination with letrozole not causing DLT in more than 33% of patients in the first treatment cycle. Twenty or more evaluable patients had to be treated before declaration of the MTD, with ≥6 evaluable patients treated at the MTD for one cycle. Criteria for evaluability were ≥21 days on alpelisib in cycle 1 or early discontinuation due to a DLT. The recommended phase II dose was defined as the highest dose at or below the MTD at which ≥75% of the patients could tolerate therapy for a minimum of 8 weeks without development of grade ≥2 hyperglycemia for more than 14 consecutive days despite initiation of oral anti-diabetic medications; and grade ≥3 rash, grade ≥2 nausea, vomiting or diarrhea, and grade ≥2 rash, all for more than 14 consecutive days of optimal medical treatment.
Safety and Radiographic Assessments
Clinical and laboratory assessments were conducted at baseline and weekly during cycle 1; on days 1 and 15 of cycle 2; and on day 1 of subsequent cycles. Safety assessments included serial electrocardiograms and fasting plasma glucose. Radiographic responses were assessed every two months using RECIST version 1.1. Best response analysis was conducted in any patient that surpassed 8 weeks of treatment, therefore having at least one set of radiographic assessments. Clinical benefit rate was defined as any patient that did not have disease progression on their radiographic assessments for a set time frame (i.e. ≥ 6 or 12 months). Patients that discontinued study treatment due to toxicity prior to their first radiographic assessment were considered non-evaluable.
Mutation Analyses
DNA was extracted using DNEasy or QiaAMP DNA tissue kits from formalin-fixed paraffin embedded (FFPE) archival tumor sections or snap frozen biopsies of metastases, respectively. Tumor cellularity was assessed by an expert breast pathologist (MES); specimens with ≥20% tumor nuclei were considered evaluable. In the few cases with paucicellular samples (<20% tumor cells), multiple sections were macro-dissected to achieve ≥20% tumor cellularity.
DNA was initially subjected to SNaPShot(18) analysis of 18 mutations in PIK3CA, PTEN and AKT1, including the common hot-spot mutations in exons 9 and 20 of PIK3CA, as previously described(19). SNaPShot is based on multiplex PCR, primer extension with fluorescently-tagged dideoxy-nucleotides and capillary electrophoresis; it is a fast, high-throughput, multiplex profiling method based on the Applied Biosystems SNaPshot platform.
Subsequently, Next Generation Sequencing (NGS) was performed using the MSK-IMPACT™ (Memorial Sloan Kettering - Integrated Mutation Profiling of Actionable Cancer Targets)(20), a hybridization capture-based assay for targeted deep sequencing of all exons and selected introns of 341 key cancer genes in DNA from FFPE tumor sections. Custom DNA probes targeting exons and selected introns of 341 genes were synthesized using the NimbleGen SeqCap EZ library custom oligo system and biotinylated to allow for sequence enrichment by capture using streptavidin-conjugated beads. Pooled libraries containing captured DNA fragments were subsequently sequenced on the Illumina HiSeq 2500 system (rapid-run mode) as 2 × 100-bp paired-end reads.
FGFR1 fluorescence in situ hybridization (FISH)
Three to four-μm tissue sections were mounted on sialinized slides and hybridized overnight with the ZytoLight SPEC FGFR1/CEN 8 Dual Color Probe (ZytoVision, Bremerhaven, Germany). Briefly, deparaffinization, pretreatment, then the slides were denatured in the presence of 10 μl probe for 6 min at 73°C and hybridized at 37°C overnight in StatSpin (Thermobrite, Abbott Molecular, Inc.). Post-hybridization SSC washes were performed at 72°C and the slides were next stained with DAPI before analysis. Slides were analyzed with Reflected light fluorescent microscope (Olympus BX60) at 100X for hot spots. Representative images of tumor cells were captured using Cytovision software. Thirty tumor cell nuclei from hot spots or random areas, were individually evaluated with the 100X by counting green FGFR1 and orange centromer 8 (CEN8) signals. The average of FGFR1 copy number and the FGFR1/CEN8 was calculated. Cases were considered as FGFR1 positive (‘amplified’) under one of the following conditions:
-
a)
The FGFR1/CEN8 ratio is ≥2.0;
-
b)
The average number of FGFR1 signals per tumor cell nucleus is ≥6;
-
c)
The percentage of tumor cells containing ≥15 FGFR1 signals or large clusters is ≥10%;
-
d)
The percentage of tumor cells containing ≥5 FGFR1 signals is ≥50%, with (a–c) representing a high-level and (d) a low-level amplification(21).
VSports注册入口 - Cell lines
MCF7 and CAMA-1 cells were obtained from ATCC and were cultured in DMEM supplemented with 10% FBS.
Lentivirus transduction
FGFR1 and GFP expressing lentiviruses were generated by co-transfecting 4 μg proviral pLX302-FGFR1 or pLX302-GFP plasmids (Open BioSystems), 3 μg psPAX2 (plasmid encoding gag, pol, rev, and Tat genes) and 1 μg pMDG2 envelope plasmid (Sigma Aldrich) into 293FT cells using Lipofectamine 2000 (Thermo Fisher). 293FT cells were fed with growth media 24 h post-transfection; virus-containing supernatants were harvested 48 and 72 h post-transfection, diluted 1:4 and applied to target cells with 8 μg/mL polybrene (Sigma Aldrich). Target cells were selected with 1 μg/mL puromycin.
Immunodetection
Cells were plated in phenol red-free IMEM + 10% dextran-charcoal-treated FBS and treated for 6 h with drugs or vehicle. Stimulation with FGF2 (Sigma Aldrich) was performed 20 min before the lysis. Lysates were generated by removing media from cells, washing monolayers with cold PBS and lysis with RIPA: 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% Deoxycholate, 0.1% SDS, 1 mM EDTA, 50 mM NaF, 1 mM NaVO4, and 1x protease inhibitor mixture (Roche). Lysates were clarified by centrifugation at 15,000 g for 15 min. Protein concentration was determined by BCA assay (Thermo scientific). For immunoblot analysis, equal amounts of protein/lane were subjected to SDS-PAGE, transferred to nitrocellulose membranes and probed with the following antibodies: phospho-Akt (Ser473), total-AKT, phospho-p44/42 MAPK (Erk1/2), total p44/p42 MAPK (Erk1/2), and actin (Cell Signaling Technologies); FGFR1 antibody was obtained from Abcam. Immunoreactive proteins were visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA).
Clonogenic Assays
For longer-term growth assays, CAMA-1 cells were seeded into six-well plates in estrogen free-media (IMEM + 10% dextran-charcoal-treated FBS) plus 5 ng/mL FGF2 and treated with vehicle, alpelisib 2 μM, lucitanib 2 μM (provided by Clovis Oncology), and the combination. Media and inhibitors were replenished every 3 days, and cells were grown for 2 weeks until the untreated wells achieved 80% confluence. Cells were fixed and stained in 20% methanol with 0.5% crystal violet and washed with water. Dried plates were imaged on a flatbed scanner. Afterwards, the crystal violet was solubilized with 20% acid acetic and quantitated by spectrophotometric detection at 490 nm using a plate reader (GloMax®-Multi Detection System, Promega).
"V体育安卓版" Cell Viability
MCF-7 cells were plated in 96-well plates (3,000 cells per well) in a volume of 100 μl of phenol red-free IMEM + 10% dextran-charcoal-treated FBS. Twenty-four h later, DMSO (vehicle) or alpelisib (0.05-4 μM) ± lucitanib 1 μM, all in the presence of 5 ng/mL FGF2 were added to the wells. After 72 h, cell proliferation was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Percentage inhibition was calculated relative to median signal from DMSO-treated wells.
"VSports最新版本" RESULTS
Study Population
Twenty-six patients were enrolled from April 2013 to February 2014 (Table 1). Overall, 85% of the patients had bone metastases and approximately 30% had visceral metastases. All patients not presenting with de novo metastatic disease had previously been exposed to an aromatase inhibitor in the adjuvant setting. In the metastatic setting, 85% had been exposed to at least one line of endocrine therapy, 69% to an aromatase inhibitor, and 31% to chemotherapy. Evaluable tumor samples were obtained in all 26 patients; 74% of these were formalin-fixed, paraffin-embedded (FFPE) blocks from the primary tumor.
Table 1.
Patient Characteristics
| Characteristics | ||||
|---|---|---|---|---|
| N = 26 | ||||
| Median age (range) | 53 | (31 – 72) | ||
| Median time since original diagnosis (months, range) | 34 | (0 – 99) | ||
| Metastatic lesions | Bone | 22 | (85%) | |
| Visceral | Liver | 7 | (27%) | |
| Lung | 6 | (23%) | ||
| Pleura/Peritoneum | 2 | (8%) | ||
| Other | Lymph nodes | 6 | (23%) | |
| Ovary | 1 | (4%) | ||
| Prior Therapies in the Metastatic Setting * | ||||
| Median number of prior therapies (range) | 2 | (1 – 4) | ||
| Endocrine therapy | Any | 22 | (85%) | |
| Aromatase inhibitor | 18 | (69%) | ||
| Chemotherapy | 8 | (31%) | ||
All patients (except for the ones presenting with de novo metastatic disease) had exposure to an aromatase inhibitor in the adjuvant setting
"V体育官网入口" Dose Escalation and MTD
Alpelisib dose was started at 300 mg/d. Dose limiting toxicity (DLT) (grade 3 rash) occurred in 2 patients at the 350 mg/d dose. These rashes eventually resolved with medical intervention (high dose anti-histamines, topical and/or oral corticosteroids) and drug interruption. One patient was able to restart at 300 mg/d. The alpelisib MTD was set at 300 mg/d; a total of 20 patients were treated at this dose (escalation and expansion phases). Less than 25% of patients required alpelisib interruption or dose reduction during the first 8 weeks of treatment.
V体育官网 - Safety Findings
Adverse events in this trial were similar to reports from the phase I single agent alpelisib trial(16) and are summarized on Table 2. The most common side effects were gastrointestinal disorders (73%), hyperglycemia (62%), fatigue (54%) and rash (42%); all of these were dose-dependent. Grade 3 adverse events associated with alpelisib were uncommon and generally cumulative (such as hyperglycemia and gastrointestinal disturbances), except for maculopapular rash, which tended to appear within the first 2 weeks of treatment. Of note, rash was less frequent in incidence and severity in patients that had been on anti-histamines (i.e., due to seasonal allergies) prior to trial enrollment. Rashes (of any grade) responded well to high dose anti-histamines (twice daily dosing), obviating the need for corticosteroids in the majority of patients developing this adverse event.
Table 2.
Most Common Adverse Events by Dose
| Toxicity Category / Toxicity (CTCAE v.4) |
300 mg/d (N = 20) | 350 mg/d (N = 6) | ||
|---|---|---|---|---|
| n (%) of subjects | ||||
| Total | Grade 3 | Total | Grade 3 | |
| Gastrointestinal disorders | ||||
| Nausea | 12 (60%) | 4 (67%) | 1 (17%) | |
| Diarrhea | 16 (80%) | 2 (10%) | 3 (50%) | |
| Mucositis (oral) | 2 (10%) | 1 (17%) | ||
| Vomiting | 5 (25%) | 2 (33%) | ||
| Dysgeusia | 4 (20%) | 3 (50%) | ||
| General disorders | ||||
| Fatigue | 9 (45%) | 5 (83%) | ||
| Investigations | ||||
| Transaminase elevation | 4 (20%) | 1 (5%) | 1 (17%) | |
| Metabolism and nutrition disorders | ||||
| Hyperglycemia | 11 (55%) | 2 (10%) | 5 (83%) | 2 (33%) |
| Anorexia | 5 (25%) | 3 (50%) | ||
| Skin and subcutaneous tissue disorders | ||||
| Rash maculopapular | 9 (45%) | 2 (33%) | 2 (33%) | |
Radiographic Assessments
Twenty-three of 26 patients (88%) were evaluable for best response, 3/26 patients discontinued treatment due to toxicity prior to their first radiographic assessment (Table 3). Five patients (19%) achieved a partial response; four of these patients had ER+/PIK3CA mutated breast cancer. Clinical benefit rate (lack of disease progression ≥6 months) was seen in 9 patients (35%; 95% CI [17%; 56%]), and 8 patients (31%; 95% CI [14%; 52]) continued to have clinical benefit for more than 12 months. Of those 8 patients, 6 had ER+/PIK3CA mutant breast cancer, and all had previous exposure to an aromatase inhibitor in the metastatic setting (Fig. 1). Five patients are still on study for more than 12 months (data cut-off June 2015).
Table 3.
Clinical Activity* by PIK3CA Mutation Status.
| Clinical Activity* | PIK3CA Mutation Status | Total | ||
|---|---|---|---|---|
| Mutated | Wild-type | |||
|
| ||||
| 16 | 10 | 26 | ||
| Partial Response | 4 (25%) | 1 (10%) | 5 (19%) | |
|
| ||||
| Stable Disease | 6 (38%) | 6 (60%) | 12 (46%) | |
|
| ||||
| Disease Progression | 4 (25%) | 2 (20%) | 6 (23%) | |
|
| ||||
|
Clinical Benefit
(no disease progression) |
≥ 6 months | 7 (44%) | 2 (20%) | 9 (35%) |
|
| ||||
| ≥ 12 months | 6 (38%) | 2 (20%) | 8 (31%) | |
|
| ||||
| Non-Evaluable | 2 (13%) | 1 (10%) | 3 (12%) | |
|
| ||||
| Still on Study | 3 (19%) | 2.(20%) | 5(19%) | |
Objective response was assessed by RECIST on the first post-baseline target lesion.
Figure 1. Treatment duration (months) with corresponding radiologic response and tumor PIK3CA mutation status.

Best response by Response Evaluation Criteria in Solid Tumors (RECIST v1.1) is depicted in yellow (partial response), blue (stable disease) and black (progression of disease) shading. Red shading indicates discontinuation of treatment due to toxicity or withdrawal. Textured bars reflect patients treated with 350 mg/d of alpelisib; solid bars reflect the ones on 300 mg/d. Arrows indicate patients that are still on study. PIK3CA mutations by NGS (in black) or SNaPShot (in pink) are shown on the vertical axis; Ø represents absence of PIK3CA mutation. Mutations on exons 9 and 20 (hot spots) were the most prevalent ones; exon 20 (H1047R) mutations appear to be associated with more clinical durable responses, in contrast to exon 9 mutations (E545K).
Mutation Analyses
Seventy-five percent (61%) of analyzed tumors were FFPE sections from the original breast cancer diagnosis (median time since original diagnosis 34 months; ranging from 0-99). We initially screened for PIK3CA mutations with SNaPShot(18) (on all 26 tumors’ DNA). We then followed with targeted capture next generation sequencing (NGS; 23 tumors were evaluable for this analysis) in order to identify somatic alterations associated with clinical benefit or lack thereof. In all cases, findings by SNaPShot were confirmed by NGS. NGS analysis showed fewer genomic alterations in tumors from patients with clinical benefit compared to those without clinical benefit (41 vs. 79 genomic alterations; not shown). Most common alterations (>10%) were PI3KCA, TP53, GATA3 and BRCA1/2 mutations, MCL1, FGFR1, CCND1, FGF3/4/19 and CCND1 amplifications, and deletions/truncations in PTEN (Fig. 2A). The majority of PIK3CA mutations were in exons 9 and 20 (E545K, E542K and Q546K, 7 tumors; H1047R alone in 5 tumors). Interestingly, 4/5 (80%) patients with tumors with PIK3CAH1047R had durable clinical responses, whereas only 2/7 (28%) tumors with mutations in exon 9 exhibited a clinical response in excess of 6 months. One of these tumors had a mutation in the Ras-binding domain (I273V) in addition to E545K. A tumor with a novel deletion in the C2 domain of PIK3CA (P447_L455del) exhibited a solid partial response in liver metastases (Fig. 1).
Figure 2. Next generation sequencing analysis of DNA from tumors of patients evaluable for clinical response.



A, Of 341 cancer-associated genes, the somatic alterations (amplifications - red, deletions - blue, truncating - black, inframe - brown, and missense - green mutations) most commonly seen (i.e. >5% cases) are depicted here. Clinical benefit was defined as lack of progression for at least 6 months on letrozole/alpelisib. A total of 23 cases with adequate tissue for DNA extraction are depicted. Tissue origin in 9/23 (39%) tumors was a metastatic biopsy (pink), whereas 14/23 (61%) of tumors were from the primary diagnostic tumor specimen (purple). Tumors from patients with clinical benefit are depicted in green (top), and tumors from patients without clinical benefit are depicted in maroon (top). Patients that discontinued trial early (i.e., < 6 months) due to toxicity are depicted in gold as non-evaluable (top). A higher proportion of PIK3CA mutations were seen in patients with clinical benefit, and overall less genomic alterations were seen when compared to patients without clinical benefit. Two of the patients with clinical benefit were found to have both PIK3CA and ESR1 mutations in their tumor. FGFR1/2 amplification and mutation in TP53, BRCA1 and KRAS were only found in cancers from patients who did not benefit from treatment. PTEN mutations were found in both groups. B, Of patients with tumors harboring the genetic alterations depicted in the X axis, the blue bars indicate the proportion of patients with clinical benefit; red bars indicate the proportion of patients without clinical benefit. The graph reveals that most patients with PIK3CA and ESR1 mutated tumors had clinical benefit, in contrast to patients with alterations in KRAS, TP53 or FGFR1, none of which had clinical benefit. C, FGFR1 amplification was confirmed by FISH analysis in the three evaluable patients with FGFR1 amplification detected by NGS.
Seventeen of 23 patients (74%) with NGS data were evaluable for clinical benefit ≥6 months; 3 patients discontinued study treatment due to toxicity prior to their first radiographic assessment; 3 patients discontinued study treatment due to toxicity prior to their second radiographic assessment and were therefore considered non-evaluable for the NGS data/outcome analysis. Seven out of 17 patients (41%) had durable responses to the combination (Fig. 2A). Five of 7 (71%) patients with clinical benefit had a PIK3CA-mutant tumor, in contrast to 5/10 (50%) patients without clinical benefit (Fig. 2B). Two patients with tumors harboring both PIK3CA and a known activating ESR1 mutation (Y537S and D538G, respectively) in their primary tumor exhibited stable disease or partial response. Interestingly, among patients evaluable for response, all those with cancers with FGFR1 and FGFR2 amplification, and KRAS and TP53 mutations progressed on treatment. FGFR1 amplification detected by NGS was confirmed by FISH analysis (Fig. 2C).
FGFR1 overexpression blocks the effect of alpelisib of ER+/PIK3CA mutant cells
To explore whether overexpression of FGFR1 is causal to resistance to alpelisib, we first examined the effect of alpelisib in the ER+ CAMA-1 breast cancer cells, which harbor FGFR1 amplification. FISH analysis of these cells showed a FGFR1/CEN8 ratio of 2.2, consistent with FGFR1 gene amplification (Fig. 3A). Growth of CAMA-1 cells in estrogen-free medium supplemented with FGF2 was completely insensitive to alpelisib, but inhibited by the FGFR1-3 tyrosine kinase inhibitor (TKI), lucitanib (Fig. 3B). Treatment with alpelisib inhibited p-AKT but not p-ERK, whereas lucitanib inhibited p-FRS2 and p-ERK but not p-AKT (Fig. 3C). We then stably transduced ER+/PIK3CA mutant MCF7 cells with a FGFR1 expression vector. Compared to MCF7GFP control cells, FGFR1 overexpressing cells exhibited slightly higher levels of p-AKT and p-ERK (Fig. 3D). Similar to the results with CAMA-1 cells, treatment with alpelisib inhibited p-AKT but not p-ERK whereas the addition of lucitanib mainly inhibited p-ERK but not p-AKT levels. Finally, MCF7GFP cells grown as monolayers had an IC50 to alpelisib <1 μM whereas this was at least 10-fold higher in MCF7FGFR1 cells. Addition of lucitanib resensitized MCF7FGFR1 cells to the PI3Kα inhibitor (Fig. 3E). Altogether, these data suggest a causal association between FGFR1 overexpression and activation of ERK with relative resistance to alpelisib.
Figure 3. FGFR1 overexpression attenuates the antiproliferative action of alpelisib.
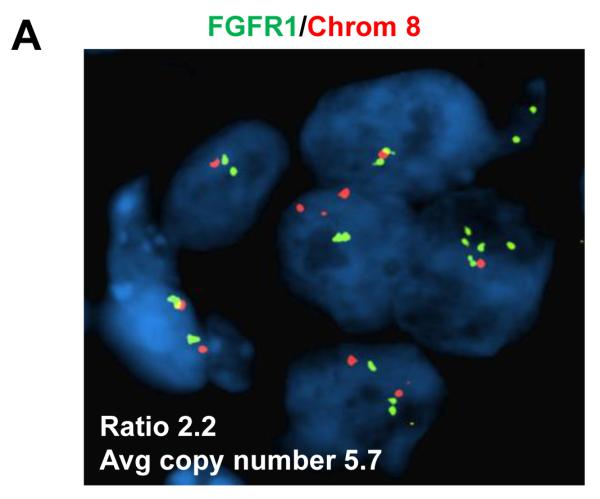
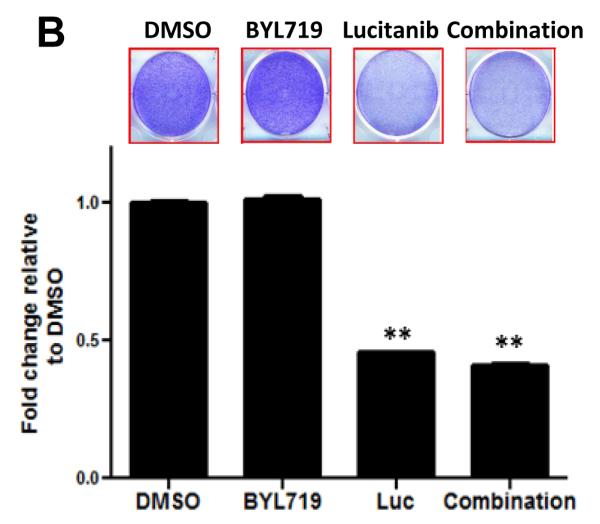
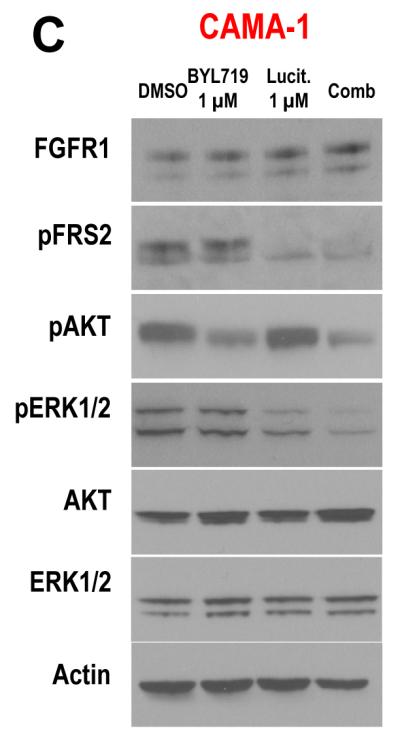


A, FGFR1 amplification was confirmed by FISH analysis in the CAMA-1 ER+ breast cancer cells as described in Methods. B, CAMA-1 cells in estrogen free-media (IMEM + 10% dextran-charcoal-treated FBS) with 5 ng/mL FGF2 were treated with 2 μM alpelisib, 2 μM lucitanib or the combination. Representative images and quantification of integrated intensity are shown (**p<0.01 vs. control, t-test). Growth of CAMA-1 cells was completely insensitive to alpelisib but inhibited by lucitanib. C, Immunoblot analysis with the indicated antibodies of lysates from CAMA-1 cells treated for 6 h with vehicle, alpelisib, lucitanib and the combination of both drugs in estrogen free-media supplemented with 5 ng/mL FGF2. Alpelisib inhibits p-AKT but not p-ERK whereas lucitanib inhibits p-ERK but not p-AKT. D, Immunoblot analysis with the indicated antibodies of lysates of MCF7GFP and MCF7FGFR1 cells that were treated for 6 h with vehicle, alpelisib, lucitanib and the combination in estrogen free-media with 5 ng/mL FGF2. Alpelisib inhibits p-AKT but not p-ERK whereas lucitanib inhibits p-ERK but not p-AKT. E, MCF7GFP and MCF7FGFR1 cells were plated in estrogen free-media and FGF2 and treated with alpelisib alone or in combination with lucitanib for 72 h. Cell proliferation was assessed by CellTiter-Glo (Promega) assay; each data point represents the mean ± SD of triplicate wells. MCF7GFP cells (black) grown as monolayers had an IC50 to alpelisib <1 μM, whereas this was at least 10-fold higher in MCF7FGFR1 cells (green). Addition of lucitanib re-sensitized MCF7FGFR1 cells to alpelisib (red).
DISCUSSION
The initial rationale for the development of isozyme-specific antagonists was to allow anti-p110α, anti-p110β and anti-p110δ agents to be delivered at maximal target-inhibitory doses while potentially avoiding the side effects of pan-PI3K inhibitors. Specific inhibitors of p110α, such as alpelisib and MLN1117, and p110β-sparing inhibitors (e.g., taselisib, GDC-0032) are being developed with a focus on PIK3CA-mutant tumors. The results from this study provide evidence that alpelisib plus letrozole is safe, tolerable and active in post-menopausal patients with ER+/HER2-negative metastatic breast cancer refractory to endocrine therapy. The MTD and recommended dose for phase II trials of alpelisib in combination with letrozole was defined as 300 mg/d. This finding was in contrast to the alpelisib single agent MTD, which is 400 mg/d(16).
Most common side effects (hyperglycemia, rash, nausea, diarrhea, and fatigue) were of similar frequency to those seen in the phase I study of single agent alpelisib(16). However, the rate of severe (grade 3) maculopapular rash was lower, most likely due to a reduced alpelisib accumulation with the 300 mg/d dose, and/or the unintended prophylactic use of anti-histaminics (i.e. due to seasonal allergies) prior to trial enrollment. In view of the urticarial-like nature of the rash, we would endorse the prophylactic use of anti-histaminics in order to minimize the incidence and severity of the alpelisib associated rash. Consistent with the hyperglycemia seen with pan-PI3K inhibitors(22-24), more than 50% of patients developed mild to moderate hyperglycemia, an on-target toxicity common to all PI3K inhibitors, especially the ones with more sustained inhibition of p110α. Severe hyperglycemia was uncommon, with none of the patients requiring administration of insulin; all of them were successfully managed with oral hypoglycemic agents such as metformin. Of note, transaminitis and gastrointestinal side effects were less frequent and severe than those observed in trials with pan-PI3K inhibitors(22-24).
The combination of letrozole and alpelisib showed sustained clinical activity: 35% of patients remained on treatment ≥6 months and 31% remained on treatment ≥12 months. The clinical benefit rate ≥6 and ≥12 months seen in patients with ER+/PIK3CA mutant metastatic breast cancer was numerically higher than in patients with wild-type PIK3CA tumors. Additionally, 4 of the 5 partial responses seen were in patients with a PIK3CA-mutant tumor. The significance of these findings is uncertain due to the small number of patients, but suggests that p110α-specific inhibitors may have greater activity against breast cancers of this genotype. The preliminary observation that the most frequent mutation in PIK3CA, H1047R, appears to be associated with higher clinical benefit from the p110α inhibitor compared to mutations in the helical domain further increases the enthusiasm for the development of PIK3CA mutant-specific inhibitors. Interestingly, a novel deletion in the C2 domain of PIK3CA (P447_L455del) was associated with a partial response. This domain is in contact with the iSH2 domain of p85, the regulatory subunit of PI3K(25). This deletion would be predicted to disrupt the interaction of p110α with p85, thus de-repressing p110α from p85-mediated inhibition. Confirmation of this hypothesis will require additional investigation beyond this report, but the partial response observed in this patient suggests this mutation is oncogenic and, thus, may confer tumor dependence on PI3K function.
Several genomic alterations were detected in addition to PIK3CA mutations, and a higher proportion of those were seen in the group of patients without clinical benefit. FGFR1 and FGFR2 amplifications and mutations in TP53, BRCA1/2 and KRAS were found only in tumors from patients that progressed on therapy. Tumors with aberrant activation of the RAS/RAF/MEK/ERK pathway, such as KRAS mutant cancers, do not respond to PI3K pathway inhibitors(26) and would not be expected to respond to alpelisib. Of note, the predominant pathway activated downstream of FGFRs has been shown to be ERK both in developmental models and in cancer cells(27, 28). Amplification of FGFR1 occurs in approximately 10% of breast cancers, predominantly in ER+ cancers where it has been associated with resistance to tamoxifen(29). Thus, to investigate whether an excess of FGFR1 is also associated with a lower response to PI3K inhibitors, we engineered ER+/PIK3CA mutant cells with FGFR1 overexpression. In these cells, the IC50 to alpelisib was increased >10-fold compared to cells without FGFR1 amplification suggesting a causal association between FGFR1 overexpression and drug resistance. Treatment with the FGFR TKI lucitanib completely restored the anti-tumor effect of alpelisib while inhibiting ERK but not AKT. Similar results were obtained with ER+/FGFR1 amplified CAMA-1 breast cancer cells, which were resistant to alpelisib but sensitive to lucitanib. These results are consistent with the lack of clinical benefit observed in the FGFR-amplified tumors in the trial herein and suggest a causal association between FGFR-overexpression and both aberrant activation of ERK and resistance to PI3K inhibitors.
Two of the patients who did not have clinical benefit had amplification of ERBB2 (HER2) in a metastatic biopsy, consistent with the notion that this genetic alteration confers resistance to endocrine therapy(30, 31). Of note, the primary tumors in these two patients were not found to have HER2-amplification by FISH. Interestingly, two patients with clinical benefit were found to have ESR1 mutations in addition to PIK3CA mutations in metastatic biopsies. These ESR1 mutations (Y537S and D538G) are in the ligand-binding domain of ERα and have previously been shown to exhibit estrogen-independent transcriptional activity(32-34). They are rarely found in primary untreated tumors and are usually detected in metastases in patients who progress after prolonged endocrine therapy. The clinical benefit seen in these patients suggests the possibility of an interaction between mutant PIK3CA and ERα that requires further study.
The results of this study demonstrate the combination of the p110α-specific inhibitor alpelisib with letrozole is safe, tolerable and active in patients with endocrine therapy-resistant ER+ advanced breast cancer, particularly those that also harbor an activating mutation in PIK3CA. Several ongoing clinical trials are now exploring the use of alpelisib and endocrine therapy in patients with both ER+/PIK3CA-mutant or wild-type breast cancer in the neoadjuvant and metastatic settings.
STATEMENT OF SIGNIFICANCE.
The combination of the PI3Kα specific inhibitor, alpelisib, and the aromatase inhibitor letrozole is safe and clinically active in patients with ER+/PIK3CA-mutant breast cancer refractory to primary endocrine therapy. This result suggests a novel molecularly targeted strategy that may abrogate or delay the emergence of antiestrogen resistance in hormone-dependent breast cancer.
ACKNOWLEDGEMENTS
Violeta Sánchez was instrumental in the tissue handling and processing at the VICC. Rajmohan Murali and Nancy Bouvier were instrumental in the tissue handling, processing, and molecular analysis. We thank the patients for their participation, and their families for support throughout the study.
Research Support: This work was funded by Stand Up to Cancer Dream Team Translational Research Grant, Program of the Entertainment Industry Foundation (SU2C-AACR-DT0209), Breast SPORE P50 CA098131, VICC Support Grant P30 CA68485, a Breast Cancer Research Foundation grant (CLA), Susan G. Komen for the Cure Foundation SAC grant (SAC100013), K23 CA127469-01A2 (IAM), and by R01 GM041890 (LCC). This study was sponsored by Novartis Pharmaceuticals.
Footnotes
Clinical Trial Information: NCT01791478
Previous Presentations: Presented in part at the American Association for Cancer Research (AACR) Annual Meeting, Philadelphia, PA, 2015
CONFLICT OF INTERESTS
Novartis ad hoc consultation: Carlos L. Arteaga, Vandana G. Abramson, Lewis C. Cantley, Ingrid A. Mayer
Novartis funding for clinical trials: Ingrid A. Mayer
AUTHOR CONTRIBUTIONS
Conception and design: Ingrid A. Mayer, Eric Winer, Carlos L. Arteaga, Yisheng Li, Lewis C. Cantley
Provision of study materials or patients: Ingrid A. Mayer, Vandana G. Abramson, Dejan Juric, Eric Winer, Carlos L. Arteaga
Collection and assembly of data: Ingrid A. Mayer, Monica V. Estrada, Melinda E. Sanders, Luigi Formisano, Justin M. Balko, David Solit, Michael F. Berger, Helen H. Hon, Yisheng Li
Data analysis and interpretation/ Manuscript writing/Final approval of the manuscript: All authors
REFERENCES
- 1.Samuels Y, Diaz LA, Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7(6):561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 3.Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010;29(37):5193–203. doi: 10.1038/onc.2010.257. [DOI (VSports app下载)] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–84. doi: 10.1158/1078-0432.CCR-06-1609. [DOI] [PubMed] [Google Scholar]
- 6.Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A. 2010;107(22):10208–13. doi: 10.1073/pnas.0907011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–61. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–13. doi: 10.1172/JCI41680. [DOI (V体育安卓版)] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69(9):3955–62. doi: 10.1158/0008-5472.CAN-08-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer discovery. 2011;1(4):338–51. doi: 10.1158/2159-8290.CD-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 13.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24. doi: 10.1038/nrc3860. ["VSports最新版本" DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–8. doi: 10.1016/j.bmcl.2013.05.007. ["VSports手机版" DOI] [PubMed] [Google Scholar]
- 15.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–29. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 16.Juric D, Gonzalez-Angulo A, Burris H, Schuler M, Schellens J, Berlin J, et al. Preliminary safety, pharmacokinetics and anti-tumor activity of BYL719, an alpha-specific PI3K inhibitor in combination with fulvestrant: Results from a phase I study. Cancer Res. 2013;73:P2-16-14. ["V体育2025版" Google Scholar]
- 17.Munster P, Hamilton E, Franklin C, Bhansali S, Wan K, Hewes B, et al. Phase lb study of LEE011 and BYL719 in combination with letrozole in estrogen receptor-positive, HER2-negative breast cancer (ER+, HER2− BC) J Clin Oncol. 2014;32:5s. (suppl; abstr 533) [Google Scholar]
- 18.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–58. doi: 10.1002/emmm.201000070. [VSports手机版 - DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abramson VG, Cooper Lloyd M, Ballinger T, Sanders ME, Du L, Lai D, et al. Characterization of breast cancers with PI3K mutations in an academic practice setting using SNaPshot profiling. Breast Cancer Res Treat. 2014;145(2):389–99. doi: 10.1007/s10549-014-2945-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17(3):251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schildhaus HU, Heukamp LC, Merkelbach-Bruse S, Riesner K, Schmitz K, Binot E, et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25(11):1473–80. doi: 10.1038/modpathol.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, et al. Stand up to cancer phase ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014;32(12):1202–9. doi: 10.1200/JCO.2013.54.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krop IE, Johnston S, Mayer IA, Dickler M, Ganju V, Forero-Torres A, et al. The FERGI phase II study of the PI3K inhibitor pictilisib (GDC-0941) plus fulvestrant vs fulvestrant plus placebo in patients with ER+, aromatase inhibitor (AI)-resistant advanced or metastatic breast cancer – Part I results. San Antonio Breast Cancer Symposium. 2014 Abstract S2-02. [Google Scholar]
- 24.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 25.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA) Proc Natl Acad Sci U S A. 2012;109(38):15259–64. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corson LB, Yamanaka Y, Lai KM, Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130(19):4527–37. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- 28.Tomlinson DC, Lamont FR, Shnyder SD, Knowles MA. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res. 2009;69(11):4613–20. doi: 10.1158/0008-5472.CAN-08-2816. ["VSports" DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29. doi: 10.1038/nrc2780. ["VSports app下载" DOI] [PubMed] [Google Scholar]
- 30.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2(2):101–12. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 31.Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15(5):261–75. doi: 10.1038/nrc3920. [DOI] [PubMed] [Google Scholar]
- 32.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73(23):6856–64. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 33.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–45. doi: 10.1038/ng.2822. [VSports手机版 - DOI] [PMC free article] [PubMed] [Google Scholar]